ⓘ Inhaltsverzeichnis dieser Seite
Was ist eine Essentielle Thrombozythämie (ET) und wie häufig tritt sie auf?
Die Essentielle Thrombozythämie (ET) gehört zu den seltenen, chronischen Myeloproliferativen Neoplasien (MPN), Erkrankungen des Knochenmarks. Diese MPN weisen viele Gemeinsamkeiten auf und lassen sich insbesondere im Anfangsstadium manchmal nur schwer voneinander unterscheiden.
Bei ET kommt es ohne erkennbare Ursache zu einer Überproduktion von Blutplättchen (Thrombozyten). Dadurch wird im Blut dauerhaft die Anzahl der Thrombozyten erhöht.
. Knochenmarkhistologie: Vermehrung mäßig polymorpher Megakaryozyten, die z. T. in Gruppen zusammenliegen (Giemsa-Färbung). Bildquelle T. Haferlach")
Thrombozyten sind für die Blutstillung zuständig. Bei Gefäßverletzungen verklumpen sie miteinander und dichten so das Blutgefäß ab. Darüber hinaus setzen sie Botenstoffe frei, die ebenfalls zur Reparatur des Gefäßschadens beitragen.
Eine zu große Anzahl von Thrombozyten im Blut erhöht die Gefahr von Thrombosen (Blutgerinnseln), dadurch steigt das Risiko für Durchblutungsstörungen, die bis zum Gefäßverschluss führen können. Wenn die Anzahl der Thrombozyten zu hoch ist, steigt das Blutungsrisiko, da die Thrombozyten sich dann gegenseitig behindern können. Der normale Referenzbereich für Thrombozyten im Blut liegt ungefähr zwischen 150.000 und 400.000 pro Mikroliter (µl), je nach Labor werden leicht abweichende Referenzbereiche angegeben.
Zählt eine ET zu den Krebserkrankungen?
Die ET gehört gemäß der aktuellen WHO-Klassifikation zu den sogenannten BCR-ABL1-negativen Myeloproliferativen Neoplasien (MPN), einer Gruppe von selbstständig wachsenden, also bösartigen, Erkrankungen. Manchmal werden die MPN auch „chronischer Blutkrebs“ genannt, was jedoch keine sehr gute Bezeichnung ist. Die ET ist keine Leukämie, aber aufgrund der unkontrollierten Zellbildung eine den Leukämien verwandte Erkrankung. Wie bei allen MPN können auch bei einer ET gesunde und krankhaft veränderte Stammzellen mit ihren jeweiligen Nachkommen lange Zeit nebeneinander existieren, wobei der Anteil an mutierten Zellen bei den einzelnen Betroffenen und in den verschiedenen Stadien der Erkrankung sehr unterschiedlich sein kann. Die ET kann sehr unterschiedlich verlaufen und hängt stark von individuellen Risikofaktoren ab.
Die ET birgt ein gewisses Risiko (überwiegend nach Jahren oder Jahrzehnten), in eine post-ET-Myelofibrose oder in eine Akute Myeloische Leukämie (AML), auch Blastenphase genannt, überzugehen.
Unterscheidung von anderen Formen der MPN
Weil die MPN viele Gemeinsamkeiten aufweisen, lassen sie sich – vor allem im Anfangsstadium – häufig nur schwer voneinander unterscheiden. Das gilt insbesondere für die Unterscheidung zwischen der ET und der präfibrotischen Myelofibrose (präPMF). Oft zeigt sich erst im Verlauf, welcher MPN-Subtyp vorliegt. Übergänge einer wahren ET in eine sekundäre Myelofibrose (post-ET-MF) oder in eine Akute Myeloische Leukämie (AML) sind selten. Vereinzelt kommt es bei JAK2-V617F-positiven Patienten zu einem Übergang in die Polycythaemia vera (PV).
Häufigkeit
Pro Jahr erkranken in Europa ca. 0,38-1,7 von 100.000 Personen neu an ET. Insgesamt erkrankt sind ca. 4-24 pro 100.000 Einwohner. Damit gehört die ET zu den sogenannten seltenen Erkrankungen (engl. orphan diseases). Das erklärt, warum viele niedergelassene Behandelnde die Erkrankung und ihren Verlauf nicht kennen und warum selbst in fachärztlichen (hämatologischen) Praxen häufig nur sehr wenige Betroffene betreut werden.
Da die ET im Allgemeinen im höheren Lebensalter auftritt, ist die Mehrheit der Erkrankten bei Diagnosestellung älter als 50 Jahre, Frauen erkranken etwa doppelt so häufig als Männer.
Ca. 20 % der Betroffenen sind jedoch bei Diagnose unter 40 Jahren. Nach einer Studie der Mayo-Clinic haben sie häufig höhere Thrombozytenwerte, eine größere Milz, häufiger eine Calreticulin-Mutation und seltener arterielle Komplikationen. Die Diagnose wird zunehmend in einem früheren Lebensalter gestellt, da sich die Erkennung der ET, insbesondere durch die aktuellen spezifischen genetischen Bluttests, verbessert hat.
Was sind die Ursachen der ET?
Die genauen Ursachen der ET sind bisher nicht bekannt. Es gibt aktuell keine gesicherten Erkenntnisse darüber, ob beispielsweise ein bestimmter Lebenswandel, der berufsbedingte Umgang mit Chemikalien, z. B. Haarfärbemittel, oder sonstige Umwelteinflüsse die Entstehung einer ET begünstigen können.
Man geht davon aus, dass sie im Laufe des Lebens durch zufällig erworbene Mutationen in blutbildenden (hämatopoetischen) Stammzellen hervorgerufen werden kann. Neueste Untersuchungen deuten sogar darauf hin, dass die Mutationen bereits im Embryonalstadium oder Säuglingsalter auftreten können und dass es viele Jahre dauert, bis die Erkrankung klinisch sichtbar wird. Diese Mutationen führen dazu, dass betroffene Stammzellen ihre normale Funktion nicht mehr erfüllen können (häufigste Mutationen bei der ET sind die JAK2V617F- und die Calreticulin-Genmutation).
JAK2-Genmutation
50 – 60 Prozent der ET-Erkrankten weisen die JAK2V617F-Mutation auf (JAK = Janus-Kinase). In Zellen, die die Mutation tragen, ist ein Enzym, die sog. Janus-Kinase-2, dauerhaft aktiviert, der „Schalter“ steht gewissermaßen permanent auf „An“. In der Folge teilt sich die Zelle häufiger als normal, was im Falle der ET zu einer unkontrollierten Vermehrung insbesondere der Thrombozyten führt.
CALRETICULIN
Bei 25 – 35 Prozent der Betroffenen von ET lässt sich eine Calreticulin- (CALR-)Mutation nachweisen. Auch diese führt zu einer Überaktivierung der JAK2-Kinase. Das Vorhandensein der CALR-Mutation hat Konsequenzen für die Behandlung und Prognose von Patienten mit ET, da sie sich häufig mit höheren Thrombozytenzahlen aber geringerem Thromboserisiko zeigt.
Bei ca. drei Prozent der Menschen mit ET liegt eine MPL-Mutation vor, eine Mutation im Gen für den Thrombopoetin-Rezeptor MPL. Diese Mutation führt, wie JAK2V617F- und CALR-Mutationen, zu einer dauerhaften Wachstumsstimulation der betroffenen Blutstammzelle. Thrombopoetin ist ein Hormon, das entscheidend an der Bildung von Blutplättchen beteiligt ist. Daher sind auch bei der MPL-Mutation die Thrombozytenzahlen erhöht.
Treibermutationen
Da die oben genannten Mutationen die „treibende Kraft“ für die Entwicklung dieser Erkrankung sind, werden sie auch als Treibermutationen bezeichnet.
Die Art der Mutation wirkt sich auf das Erscheinungsbild und die Prognose der ET aus. So haben Betroffene mit JAK2V617F-Mutation ein höheres Thromboserisiko als jene mit einer CALR-Mutation, obwohl diese häufig höhere Thrombozytenzahlen aufweisen.
Triple negative
Erforschung weiterer Mutationen
Zusätzlich zu den Treibermutationen (JAK2V617F, CALR und MPL) können bei allen MPN zusätzliche, sogenannte „passenger mutations“ (Passagier-Mutationen) eine Rolle spielen, die nicht die MPN-Erkrankung auslösen, aber deren Verlauf beeinflussen können. Sie werden oft bei anderen Knochenmarkerkrankungen gefunden. Einzelne solche Mutationen, wie eine ASXL1-Mutation, können einen ungünstigen Einfluss auf die Prognose haben, unabhängig davon, welche Grunderkrankung vorliegt. Andere Mutationen, die den Verlauf der ET negativ beeinflussen können, sind SH2B3, SRSF2, U2AF1, TP53, IDH2 und EZH2.
Ist ET vererbbar?
Nach dem jetzigen Stand der Forschung sind die MPN, also auch die ET, in der Regel nicht genetisch vererbt. Stammbaumuntersuchungen deuten jedoch darauf hin, dass die MPN in Einzelfällen familiär gehäuft auftreten können. Eine genetische Veranlagung, mit der die Wahrscheinlichkeit steigt, die Krankheit im Laufe des Lebens zu erwerben, ist in einem sehr geringen Prozentsatz von Fällen nachgewiesen worden. Bei familiären Häufungen von MPN über mehrere (mindestens drei) Generationen oder Erkrankungen an anderen hämatologischen Neoplasien oder weiteren Krebserkrankungen wird gemäß Leitlinien der DGHO eine humangenetische Beratung empfohlen.
Welche Symptome können auf eine ET hindeuten?
Die Mehrzahl der ET-Betroffenen lebt über lange Zeit weitgehend beschwerdefrei. Zwischen dem Auftreten erster Symptome bis zur korrekten Diagnosestellung liegt häufig nochmals ein längerer Zeitraum.
Wenn Symptome auftreten, sind dies Beschwerden, die oft erst im Rückblick mit einer ET in Verbindung gebracht werden.
Bei längerer Erkrankungsdauer kann die Milz vergrößert sein.
Häufige Symptome sind sogenannte Mikrozirkulationsstörungen. Zum Beispiel Durchblutungsstörungen in kleinen Gefäßen (mikros=klein) an Händen und/oder Füßen oder im Gehirn (Schwindel, Kopfschmerzen, Sehstörungen/Flimmersehen und Sprachstörungen). Die meisten dieser Beschwerden können sich auch unabhängig von der Erkrankung zeigen und werden häufig erst im Rückblick mit der ET in Verbindung gebracht. Daher führen oft erst schwerwiegende Komplikationen wie venöse oder arterielle Thrombosen zur Diagnose der ET.
- Müdigkeit, Abgeschlagenheit
- Ohrensausen, Tinnitus (Ohrgeräusche)
- Kopfschmerzen / Migräne
- Schwindel
- Wadenkrämpfe
- Schmerzen in den Beinen
- Blutergüsse (Hämatome), teilweise mit Verhärtungen/Schwellungen, die sich nur langsam zurückbilden
- Nasenbluten
- Zahnfleischbluten
- ungewöhnlich starke oder schwache Menstruationsblutungen
- schmerzhafte Rötungen, Schwellungen, Kribbeln, Brennen oder Taubheitsgefühle in den Finger- und/oder Zehenspitzen (Erythromelalgie)
- Missempfindungen der Haut (z. B. das Gefühl, als würde etwas über den Arm krabbeln)
- Flimmerskotome
Diagnose – Wie wird die ET nachgewiesen?
Die ET wird häufig als Zufallsbefund durch die im Rahmen einer Blutentnahme nachgewiesenen hohen Thrombozytenzahlen diagnostiziert. Diese können aber auch andere Ursachen haben.
Folgende Szenarien können zur Diagnose einer ET führen:
- Bei einer Routineuntersuchung oder im Rahmen der Diagnostik und Therapie anderer Erkrankungen fallen dauerhaft erhöhte Thrombozyten auf.
- Betroffene suchen eine ärztliche Praxis auf, weil sie unter Durchblutungsstörungen (Mikrozirkulationsstörungen) oder einer nicht erklärbaren chronischen Erschöpfung mit andauernder Müdigkeit, sogenannte Fatigue, leiden. Diese erkrankungsbedingte dauerhafte Erschöpfung wird inzwischen von MPN-Fachleuten als wichtiges Kriterium in die Diagnostik einbezogen.
- Schwerwiegende Komplikationen wie eine Thrombose, ein Herzinfarkt oder ein Schlaganfall führen zur Diagnose ET.
Die Erfahrungen der Mitglieder des MPN-Netzwerks zeigen, dass viele Behandelnde die vielschichtige Gesamtsymptomatik zunächst nicht richtig einordnen, da Veränderungen des Blutbildes verschiedene Ursachen haben können. Mitunter werden sie sogar als Laborfehler eingestuft und ignoriert. Außerdem ist die ET – wie bereits erwähnt – noch immer relativ unbekannt. Eine Erhöhung der Thrombozytenwerte kann auch andere Ursachen haben, z.B. sekundäre Thrombozythämie oder reaktive Thrombozytose. Dies wird zunächst abgeklärt, bevor eine Essentielle Thrombozythämie in Erwägung gezogen wird.
Wichtig ist, dass von ärztlicher Seite frühzeitig an die Möglichkeit einer ET gedacht wird, vor allem dann, wenn die Werte über einen längeren Zeitraum erhöht bleiben. Bei dauerhaft erhöhten Thrombozyten über 450.000 / µl Blut, ohne dass sich eine eindeutige Ursache dafür finden lässt, sollte man baldmöglichst fachärztlichen Rat einholen, um sicher zu gehen, dass eine Diagnose auf Basis der aktuellen Behandlungsleitlinien der Deutschen Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO) gestellt wird.
Die ET-Diagnosekriterien im Überblick (nach WHO 2022)
Hauptkriterien
- Dauerhafte Thrombozytenzahl von > 450 000 / µl im peripheren Blut
- Knochenmarkhistologie:
- Vermehrung der Megakaryozytenlinie mit erhöhter Zahl vergrößerter, reifer und hyperlobulierter (hirschgeweihartiger) Megakaryozyten
- keine Erhöhung oder Linksverschiebung der Granulopoese oder Erythropoese
- keine oder nur geringe Zunahme (Grad 0-1) der Retikulinfasern
- WHO-Kriterien für eine BCR-ABL1-positive Chronische Myeloische Leukämie (CML), eine PV, PMF oder andere MPN sind nicht erfüllt
- Nachweis einer JAK2-, CALR- oder MPL-Mutation
Nebenkriterien
- Vorkommen eines anderen klonalen Markers oder fehlende Hinweise auf eine reaktive Thrombozytose
Die Diagnose ET gilt als gesichert, wenn entweder alle vier Hauptkriterien oder die ersten drei Hauptkriterien und das Nebenkriterium erfüllt sind.
Welche Untersuchungen werden bei der Diagnose durchgeführt?
Blutuntersuchung
Die Laboranalyse der Blutprobe zeigt bei fast allen ET-Erkrankten erhöhte Thrombozytenwerte, allerdings kann das Ausmaß der Thrombozytenerhöhung unterschiedlich sein.
Ultraschall des Bauchraums (Abdominelle Sonografie)
Als Folge der ET ist bei den meisten Betroffenen die Milz mehr oder weniger stark vergrößert (Splenomegalie). Grund ist der krankheitsbedingt gesteigerte Zellumsatz, weshalb die Milz besonders viele alte und veränderte Blutzellen abbauen muss. Üblicherweise treten erst bei einer stark vergrößerten Milz Bauchbeschwerden auf. In den meisten Fällen merken die Betroffenen nichts von der Milzvergrößerung, und diese fällt erst beim Ultraschall auf.
Molekulargenetische Untersuchung
Der Nachweis einer der drei Treibermutationen JAK2, CALR und MPL wird in einer molekulargenetischen Untersuchung im Rahmen einer Blutuntersuchung bestimmt. Einer der Tests fällt bei etwa 90 Prozent der ET-Betroffenen positiv aus. 10 – 12 % der ET-Erkrankten weisen keine Treibermutation auf, sind also triple-negativ. Das Fehlen der drei Treibermutationen schließt also eine MPN nicht aus.
Außer dem Nachweis einer Mutation ist die Knochenmarkpunktion (KMP) die wichtigste Methode, um eine ET eindeutig diagnostizieren zu können – und daher meist unumgänglich. Die veränderten blutbildenden Zellen und die Architektur des Knochenmarks lassen sich nur mittels Knochenmarkzytologie und -histologie unter dem Mikroskop exakt untersuchen und nur so der MPN-Subtyp genau einordnen.
Wie oben erwähnt, ist im Speziellen die Abgrenzung der ET zur präfibrotischen PMF (präPMF) wichtig. Diese wurde im Jahre 2016 von der WHO als eigener Subtyp innerhalb der MPN eingeführt. Betroffene mit präPMF werden zuweilen als ET-Erkrankte eingestuft, haben aber im Vergleich zur ET einen etwas ungünstigeren Verlauf. Eine Abgrenzung ist auch im Hinblick auf eine adäquate Behandlung wichtig.
Typischer KMP-Befund bei einer ET
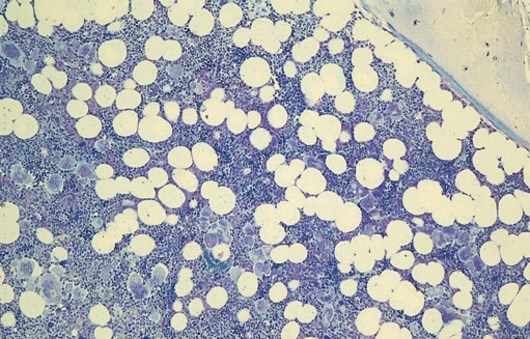
Die Ergebnisse der Knochenmarkuntersuchung stellen die einzige Möglichkeit dar, die einzelnen MPN sicher voneinander zu unterscheiden und sind deshalb für die weitere Behandlung und Prognose von großer Wichtigkeit. Daher empfiehlt es sich, die Gewebeprobe ggf. zusätzlich von einem spezialisierten Referenzlabor hämatopathologisch zweitbegutachten zu lassen.
Wichtig zu wissen:
Letztlich stellt jede Knochenmarkpunktion nur eine Momentaufnahme dar, die in der Regel keine Aussage über den weiteren Verlauf der Erkrankung zulässt. Der Befund kann abhängig von der Entnahmestelle variieren.
Welche Prognose hat die ET?
Prognoseangaben, insbesondere aus dem Internet, sind vielfach veraltet und daher irreführend, und sollten mit Vorsicht betrachtet werden. Hinweise zum Umgang mit Prognoseangaben bieten die Info-Blätter der Deutschen Leukämie und Lymphom-Hilfe (DLH).
Die Lebenserwartung von ET-Betroffenen ist günstig, wenn die Erkrankung präzise diagnostiziert wurde und eine fachgerechte Behandlung erfolgt. Hauptrisiko stellen thromboembolische Ereignisse dar. Dieses Risiko lässt sich jedoch mit einer geeigneten Therapie deutlich reduzieren. Die Behandlung der ET richtet sich danach, wie groß das individuelle Risiko einer thromboembolischen Komplikation ist.
Um das Risiko für ein thromboembolisches Ereignis abzuschätzen, wurden unterschiedliche Risikogruppen definiert. Noch findet meist der konventionelle Risikoscore Anwendung.
Risikogruppen der ET (konventioneller Risikoscore)
Hochrisiko (einer oder mehrere der folgenden Faktoren treffen zu)
- Biologisches Alter > 60 Jahre
- Vorangegangene thromboembolische Ereignisse oder Blutungskomplikationen
- Thrombozytenzahl > 1 500 000/µl
Intermediärrisiko (bei ET-unabhängigen vaskulären Risikofaktoren)
- Vorliegen von Herz-Kreislauf-Risikofaktoren wie Bluthochdruck, Diabetes mellitus, erhöhte Cholesterinwerte, Rauchen, Übergewicht, Gefäßerkrankungen, hormonelle Verhütung
- Erhöhte Thrombophiliemarker (angeborene, durch eine Blutuntersuchung feststellbare Faktoren, die auf eine erhöhte Blutgerinnungsneigung hinweisen)
Niedrigrisiko
- Keine der oben genannten Risikofaktoren
Die Entscheidung, ob eine Therapie empfohlen wird, und welche Therapie dies ist, basiert auf den obigen Risikoeinteilungen. Daneben gibt es neuere Prognose-Bewertungssysteme wie den IPSET-Score, die auch molekulare Marker einbeziehen. Allerdings werden diese neuen Prognosescores bisher in Deutschland noch nicht flächendeckend für Therapieentscheidungen angewendet.
Die Erfahrungen der Mitglieder des MPN-Netzwerks zeigen, dass schlecht informierte Behandelnde manchmal schockierende Angaben zur Lebenserwartung machen, die Betroffene stark verunsichern und unnötig Ängste schüren. Wird die Erkrankung aufmerksam beobachtet und bei Bedarf behandelt, haben Betroffene eine nahezu normale Lebenserwartung.
Wie ist der Verlauf einer ET?
Das Kennzeichen aller Myeloproliferativen Neoplasien ist, zumindest am Anfang, eine gesteigerte Produktion (Proliferation) bestimmter Blutzellen. Bei der ET steht nur eine Zellreihe im Vordergrund: die Thrombozyten. Im Knochenmark sind die Vorläufer dieser Thrombozyten, die Megakaryozyten, gesteigert, welche die Thrombozyten ins Blut abgeben. In den meisten pathologischen Befundberichten ist bei einer ET daher häufig von einer gesteigerten Megakaryopoese die Rede. Eine Auslagerung der Blutbildung aus dem Knochenmark in andere Organe findet zu diesem frühen Zeitpunkt der Erkrankung in der Regel noch nicht oder nur begrenzt statt. Die Milz ist daher im frühen Stadium nicht oder nur geringgradig vergrößert. Die bei Gesunden etwa 4 × 7 × 11 Zentimeter große Milz (Volumen 26-250 ml) kann auf weit über 25 Zentimeter Länge anschwellen und große Teile des Bauchraumes ausfüllen. Dies hat zur Folge, dass Organe wie der Magen verdrängt und in ihrer Funktion beeinträchtigt werden und es kann zu linksseitigen Oberbauchbeschwerden kommen.
Der Übergang einer durch eine Knochenmarkpunktion und einen histologischen Befund bestätigten „wahren“ ET in eine Myelofibrose (post-ET-MF) ist sehr selten. Häufig handelt es sich bei diesen vermeintlichen Übergängen um eine präPMF, die fälschlicherweise als ET diagnostiziert wurde. Die präPMF wurde 2016 neu in die WHO-Klassifikation aufgenommen und bis dahin häufig als ET diagnostiziert.
Vereinzelt kommt es bei JAK2-V617F-positiven Erkrankten zu einem Übergang in eine Polycythaemia vera (PV). Oft kündigt sich dieser Übergang durch einen neu aufgetretenen erhöhten Hämatokritwert im Blut oder neu aufgetretene Symptome wie aquagener Pruritus (Juckreiz) nach Kontakt mit Wasser) oder andere Symptome an.
Der Übergang in eine Akute Myeloische Leukämie (AML) ist bei der ET eher selten.
Für die weitere Verlaufseinschätzung ist es daher sehr wichtig zu kontrollieren, ob es sich wirklich um eine wahre ET handelt. Auch aus diesem Grund ist es ratsam, den individuellen Krankheitsverlauf kontinuierlich ärztlich begleiten zu lassen – am besten in einem auf die Behandlung von MPN spezialisierten Zentrum. Dort ist man in der Lage, Veränderungen im Krankheitsverlauf (Beschwerden, Blutbild, Milzgröße, etc.) rasch zu erkennen, die Vor- und Nachteile einer medikamentösen Behandlung sorgfältig abzuwägen und gemeinsam mit der betroffenen Person die optimale Therapie zu finden.
Welche Komplikationen und Risiken können bei der ET auftreten?
Was versteht man unter Komplikationen und Risiken?
Was versteht man unter einer Komplikation?
Als Komplikation bezeichnet man in der Medizin eine unerwünschte Folge einer Krankheit, eines Unfalls, eines Eingriffs oder eines Medikaments, die nicht im engeren Sinn zum Krankheitsbild gehört. Komplikationen machen meist den Beginn einer Therapie oder deren Änderung erforderlich. Die Verschlimmerung eines Krankheitszustandes wäre z.B. eine Komplikation.
Was versteht man unter einem Risiko?
Ein Risiko weist je nach Fachgebiet einen unterschiedlichen Begriffsinhalt auf. Allgemein wird hierunter die Möglichkeit des Eintritts künftiger Ereignisse, die nachteilige Auswirkungen wie Verlustgefahren in sich bergen, verstanden. Beispiel eines Risikos ist der Übergang der ET in eine post-ET-MF (Myelofibrose).
Komplikationen bei einer ET
Bei einer ET müssen nicht zwangsläufig schwerwiegende Komplikationen auftreten. Allerdings ist die statistische Wahrscheinlichkeit größer als bei Nichtbetroffenen.
Durchblutungsstörungen
Die Störung der Blutstillung und Gerinnung infolge der hohen Thrombozytenzahlen bedingt ein erhöhtes Risiko für Durchblutungsstörungen und damit einhergehend für ein thromboembolisches Ereignis, die bei der ET häufigste und eine der am meisten gefürchtete Komplikation.
Mit zunehmendem Lebensalter treten solche Ereignisse gehäuft auf, da auch der allgemeine Zustand der Gefäße für die Blutgerinnung eine Rolle spielt.
Wichtig zu wissen:
Die Höhe der Thrombozyten allein sagt nichts über das Risiko aus, bei einer ET Beschwerden oder schwerwiegende Komplikationen zu entwickeln. Die Erfahrung zeigt vielmehr, dass auch Erkrankte mit nur geringfügig erhöhten Thrombozyten Symptome zeigen können, während Betroffene mit stark erhöhten Werten nicht unbedingt Beschwerden haben müssen.
Chronischer Entzündungszustand
Außer der veränderten Anzahl der Zellen sind die MPN-Erkrankungen auch durch eine Art chronischer Entzündungszustand gekennzeichnet, der Gefäßveränderungen und damit ebenfalls eine Thromboseneigung verursachen kann.
Blutungsneigung
Paradoxerweise kann bei sehr hohen Thrombozytenzahlen (1-1,5 Million pro µl) die Blutungsneigung erhöht sein, da Thrombozyten den für die Blutgerinnung wichtigen Von-Willebrand-Faktor abbauen können. Das kann zu spontanen Haut- und Schleimhautblutungen (Blutergüsse, Nasenbluten, etc.) sowie im Einzelfall zu Magen-Darm- oder Hirnblutungen führen.
Die Einnahme von ASS (Acetylsalicylsäure, „Aspirin“) muss in diesen Fällen unbedingt hämatologisch abgeklärt werden.
Wichtig zu wissen:
Sie können das persönliche Risiko für etwaige Komplikationen senken durch:
- eine aufmerksame Selbstwahrnehmung und zwingend erforderliche regelmäßige ärztliche Kontrollen
- das Beachten der allgemeinen Empfehlungen zur Begrenzung des Herzinfarkt- und Schlaganfallrisikos:
- Nicht rauchen
- Effektive Behandlung von Herz- Kreislauferkrankungen, Diabetes, hohem Cholesterinspiegel, etc.
- Ausgewogene Ernährung (z.B. mediterran)
- Reduzierung von Übergewicht
- Regelmäßige Bewegung, z.B. Ausdauersport, Venengymnastik
- Kontrolle von Thrombophiliemarkern (Blutgerinnungsmarker)
- Ausreichende Flüssigkeitszufuhr, sofern nicht andere Krankheiten dagegen sprechen
Besondere Maßnahmen bei längeren Autofahrten und Langstreckenflügen:
- genügend trinken
- sich immer wieder bewegen
- evtl. Kompressionsstrümpfe tragen
- ggf. Heparin spritzen (sprechen Sie in einem ärztlichen Gespräch an, ob und ab welcher Länge des Fluges und in welcher Dosis dies für Sie empfohlen wird)
Risiko: Übergang in eine andere Krankheitsform
Die ET kann in eine andere Myeloproliferative Neoplasie übergehen, z. B. in eine Myelofibrose (post-ET-MF), eine Polycythaemia vera (PV) oder eine Akute Myeloische Leukämie (AML). Genaueres unter Verlauf.
Wann sollte mit einer Behandlung begonnen werden?
Die Entscheidung, wann der Zeitpunkt für einen Therapiebeginn gekommen ist, ist stets ein Kompromiss – zwischen der Notwendigkeit, krankheitsbedingte Beschwerden zu lindern und Komplikationen vorzubeugen auf der einen Seite und dem Risiko medikamentöser Nebenwirkungen auf der anderen. Für diese Entscheidung werden von ärztlicher Seite Kriterien zur Risikobewertung herangezogen. Diese berücksichtigen unterschiedliche Faktoren wie das Alter, Stadium der Erkrankung, Symptome und das persönliche Thromboserisiko, welche aber nur als Orientierung dienen, da jeder Fall individuell zu betrachten ist.
Faktoren für die Risikoabschätzung
Die DGHO-Behandlungsleitlinien unterscheiden verschiedene Risikogruppen bei Essentieller Thrombozythämie.
Niedrigrisiko
- Alter ≤ 60 Jahre
- Thrombozyten < 1,5 Mio/μl
- keine früheren Thrombosen oder schweren Blutungen
Betroffene mit Niedrigrisiko tragen gegenüber der Normalbevölkerung kein eindeutig erhöhtes Risiko für Komplikationen, weshalb der Vorteil einer medikamentösen Behandlung nicht gesichert ist. Insbesondere jüngere Erkrankte, die bisher keine Symptome zeigen, kommen – engmaschige Kontrollen vorausgesetzt – meist über lange Zeit ohne eine spezielle Therapie aus.
Intermediärrisiko
- Vorliegen von Herz-Kreislauf-Risikofaktoren (Bluthochdruck, Diabetes mellitus, erhöhte Cholesterinwerte, Rauchen) oder erhöhter Thrombophiliemarker
- Alter ≤ 60 Jahre
- Thrombozyten < 1,5 Million/μl
- keine früheren Thrombosen oder schweren Blutungen
Vor allem bei Betroffenen mit Intermediärrisiko ist es notwendig, stets den Einzelfall zu betrachten, da für dieses Risikoprofil keine eindeutigen Empfehlungen vorliegen. Der ärztliche Rat für diese Gruppe beschränkt sich im Allgemeinen auf die Einnahme von niedrig dosiertem ASS (Acetylsalicylsäure).
Hochrisiko
- Alter > 60 Jahre
- und/oder Thrombozyten ≥ 1,5 Mio./μl
- und/oder frühere Thrombosen oder schwere Blutungen
Betroffene mit Hochrisiko sollten in der Regel eine zellreduzierende (zytoreduktive) Therapie beginnen, um Komplikationen wie Thrombose, Herzinfarkt, Schlaganfall und schwere Blutungen zu verhindern. Natürlich bedeutet dies keinen 100%-igen Schutz, aber das Risiko für diese Komplikationen kann gesenkt werden.
Weitere Faktoren, die ebenfalls in die Risikoabschätzung einfließen
- Wie alt ist die Patientin? Ausschlaggebend ist vor allem das biologische Alter.
- Seit wann besteht die Diagnose und wie war der bisherige Krankheitsverlauf?
- Sind beim Patienten oder in seiner Familie bereits gravierende Komplikationen aufgetreten wie z. B. Thrombosen oder Blutungen?
- Bestehen ET-unabhängig Risikofaktoren für thromboembolische Ereignisse wie z. B. Thrombophiliemarker, Rauchen, Pille, Gefäßerkrankungen, Diabetes oder Übergewicht?
- Welche Treibermutation liegt der Erkrankung zugrunde?
- Liegt eine Milzvergrößerung vor und bereitet diese Probleme (Oberbauchschmerzen o. Ä.)?
- Wie stark sind die Thrombozyten erhöht?
- Wie ist das Gesamtbefinden? Wie häufig zeigen sich Symptome, insbesondere Mikrozirkulationsstörungen?
- Wie wird die Verträglichkeit von Medikamenten eingeschätzt? (Allergien, Autoimmunerkrankungen, Nebenwirkungen, etc.)
Wichtig zu wissen: Thrombozytenzahlen können schwanken
Schwankende Thrombozytenzahlen sind sowohl bei Gesunden als auch bei ET-Erkrankten nicht ungewöhnlich. Ursache können u.a. eine unterschiedlich starke Zellbildung im Knochenmark sein oder Ungenauigkeiten bei der Messung. Es empfiehlt sich, regelmäßige Blutuntersuchungen stets in derselben Praxis und vom selben Labor vornehmen zu lassen und darauf zu achten, dass das Blut immer auf die gleiche Weise entnommen wird (in der Regel aus einer Armvene). Daneben macht es einen Unterschied, ob die Messung bereits in der Praxis (d. h. am Ort der Blutabnahme) oder erst nach dem Transport in das Labor erfolgt.
Wie wird die ET behandelt?
Eine Heilung der ET ist nach heutigem Wissensstand durch eine medikamentöse Therapie nicht möglich.
Die derzeit einzige Chance auf Heilung von MPN ist die Stammzelltransplantation (SZT). Allerdings wird dieses Verfahren bei der ET nicht angewendet, da es immer noch mit ernsten Behandlungsrisiken (z. B. GvHD) behaftet ist, und da die Lebenserwartung von ET-Erkrankten gegenüber der Normalbevölkerung praktisch nicht eingeschränkt ist. Das Nutzen-Risiko-Verhältnis einer SZT wäre für Betroffene mit ET zu gering.
Gleichwohl kann die ET gut behandelt werden. Eine einheitliche Therapieempfehlung für alle ET-Erkrankten gibt es nicht, auch weil Ausprägung und Verlauf in der Regel sehr unterschiedlich sind. Bei der Entscheidung für eine bestimmte Art der Behandlung ist nicht nur das Gesamtbefinden der erkrankten Person und die Risikoabschätzung zu berücksichtigen, sondern auch, wie gut sie die Einnahme eines bestimmten Medikamentes voraussichtlich verträgt. Allergische Reaktionen auf einen Wirkstoff sind daher ebenso auszuschließen wie unerwünschte Wechselwirkungen mit anderen Medikamenten und möglicherweise bestehenden Vorerkrankungen. Nicht zuletzt spielt auch die persönliche Einstellung von Betroffenen zur geplanten Behandlung eine entscheidende Rolle.
Ziele der Therapie
Vorbeugen von Durchblutungsstörungen
Treten erste Symptome auf, die auf Durchblutungsstörungen hinweisen, erhalten ET-Erkrankte in der Regel zunächst niedrig dosiertes ASS (üblicherweise 100 mg pro Tag). Man beobachtet, ob die Symptome zurückgehen. Besteht eine gesteigerte Blutungsneigung, muss auf ASS jedoch verzichtet werden, da sich die Symptomatik ansonsten noch verstärken kann.
Reduzierung der Thrombozytenzahlen
Ist eine Behandlung mit ASS nicht möglich oder zeigt sie keinen Effekt, kann eine Therapie zur Reduzierung der Thrombozytenzahlen sinnvoll sein. Die Frage, welches Medikament sinnvoll, effektiv und verträglich ist, ist individuell abzuwägen und auszutesten.
Verhindern weiterer thromboembolischer Ereignisse
Tritt ein gravierendes Ereignis wie eine Thrombose, ein Herzinfarkt oder ein Schlaganfall auf, ist eine sofortige zellreduzierende Therapie angezeigt. Ziel ist es, die Thrombozytenzahlen rasch zu senken, um so die akute Gefahr abzuwenden und das Risiko weiterer Komplikationen zu minimieren.
Welche Therapieoptionen gibt es?
Beobachten und Abwarten – watch and wait
Symptomfreie Betroffene der Niedrigrisiko-Gruppe kommen in der Regel ohne medikamentöse Therapie aus. Die Behandlung beschränkt sich darauf, den Krankheitsverlauf zu beobachten durch eine etwa vierteljährliche Kontrolle des Blutbildes und abzuwarten („watch and wait“-Strategie). Empfohlen wird eine umfassende jährliche Untersuchung. Dazu gehört auch eine Sonographie der Milz. Eine erneute Knochenmarkpunktion ist nur nötig, wenn sich der Krankheitsverlauf gravierend verändert, z.B. durch das Auftreten von Erythrozyten-Tränenformen (auch als „tear drop“-Erys bezeichnet), unreifen Erythrozyten (Erythroblasten) oder unreifen weißen Blutkörperchen im Blut, die den beginnenden Übergang in eine Myelofibrose anzeigen können.
Thrombozytenaggregationshemmer
Thrombozytenaggregationshemmer (TAH) verhindern die Verklumpung der Blutplättchen und setzen so die Blutstillungs- und Gerinnungsaktivität herab. Dies verbessert die Fließeigenschaften des Blutes und beugt Durchblutungsstörungen vor.
Acetylsalicylsäure (ASS)
Meist wird bei ET der Wirkstoff Acetylsalicylsäure (ASS) in niedriger Dosierung verordnet. ASS verbessert in der Regel Mikrozirkulationsstörungen, die sich als Durchblutungsstörungen an Händen und Füßen, Schwindel, Kopfschmerzen oder Sehstörungen bemerkbar machen können. In der Referenzstudie (ECLAP-Studie bei PV) erwies sich die Einnahme von ASS als wirkungsvoller als keine ASS-Therapie, da sie das Auftreten von arteriellen und schweren venösen Thrombosen reduzierte, weshalb sie grundsätzlich allen PV-Betroffenen empfohlen wird, bei denen medizinisch nichts gegen eine Einnahme spricht (keine Kontraindikationen). Ebenso wird sie allen ET-Betroffenen mit Intermediärrisiko und Hochrisiko empfohlen. Randomisierte klinische Studien, die eindeutig belegen, dass eine ASS-Therapie bei ET-Erkrankten besser ist als keine ASS-Therapie, gibt es allerdings bisher nicht.
Wichtig zu wissen:
ASS ist in Deutschland in einer Standarddosierung von 500mg pro Tablette als frei verkäufliches Schmerzmittel erhältlich und wird oft unbedacht angewendet. Da der Wirkstoff die Blutungsneigung verstärkt, sollte die Einnahme nicht ohne ärztliche Zustimmung erfolgen.
Gegenanzeigen (Kontraindikationen)
Eine relative Gegenanzeige für ASS besteht immer dann, wenn eine erhöhte Blutungsneigung besteht oder Betroffene unter Magen- und Darmgeschwüren leiden.
Eine sehr hohe Thrombozytenzahl von mehr als 1 bis 1,5 Mio./µl spricht ebenfalls gegen die Einnahme von ASS. In diesen Fällen liegt häufig ein erworbenes von-Willebrand-Syndrom vor und damit eine erhöhte Blutungsneigung, weshalb ASS nur in begründeten Ausnahmefällen, bzw. erst nach Absenkung der Thrombozytenzahl zum Einsatz kommen darf. In dieser Situation wird empfohlen, die von-Willebrand-Faktor-Aktivität zu bestimmen: Liegt diese über 30 Prozent, kann ASS im Allgemeinen eingenommen werden.
Alternativen
Manche Menschen leiden an einer Unverträglichkeit oder sprechen auf ASS nicht an. Mittlerweile gibt es mit den Wirkstoffen Clopidogrel, Prasugrel oder Ticagrelor einige Alternativen, die nach ärztlicher Rücksprache angewandt werden können. Niedrig dosiertes Heparin kann ebenfalls eine Alternative darstellen.
Einnahmeform
Tabletten, i. d. R. täglich (50 -100 mg). Da jeder Mensch unterschiedlich auf ASS reagiert, kann es ggf. notwendig sein, die Dosis und den Verabreichungszyklus (z.B. nur jeden 2. Tag) individuell anzupassen. Manche Fachleute empfehlen, die Einnahme von ASS auf 2x täglich zu verteilen. Mit der magenschonenderen Variante ASS protect kann man Magenproblemen evtl. vorbeugen. Dabei ist zu beachten, dass man ASS zum Essen, die magenschonende Variante ASS protect jedoch ca. 30 Minuten vor dem Essen einnehmen soll.
Zellreduzierende Medikamente
Eine Behandlung mit zellreduzierenden Medikamenten ist immer dann die Therapie der Wahl, wenn Betroffene biologisch älter als 60 Jahre sind, es in der Vergangenheit bereits zu thromboembolischen Ereignissen gekommen ist oder die Thrombozytenzahl über 1,5 Millionen
pro µl steigt. Ziel der Behandlung ist es, die Thrombozyten in den Normbereich abzusenken. Der normale Referenzbereich für Thrombozyten im Blut liegt ungefähr zwischen 150.000 und 400.000 pro Mikroliter (µl), je nach Labor werden leicht abweichende Referenzbereiche angegeben.
Es werden verschiedene Wirkstoffe eingesetzt (u.a. Hydroxyurea, Anagrelid, Interferon), die sowohl mit ASS als auch untereinander kombinierbar sind. Um die Nebenwirkungen möglichst gering zu halten, kann es unter Umständen sinnvoller sein, zellreduzierende Medikamente niedrig zu dosieren und ASS zu nehmen, statt auf ASS um den Preis sehr hoher Dosierungen zu verzichten. Hier ist es die ärztliche Aufgabe, Risiken und Nebenwirkungen gegeneinander abzuwägen und dies mit der betroffenen Person zu besprechen.
Da die Blutwerte nach dem Absetzen der zellreduzierenden Medikamente unterschiedlich schnell wieder ansteigen, ist – außer in einzelnen Fällen nach Interferon-alpha-Behandlung – in der Regel eine dauerhafte Erhaltungstherapie erforderlich. Die Dosierungen der einzelnen Wirkstoffe sind individuell verschieden und können sich im Verlauf der Erkrankung ändern. Deshalb sind regelmäßige Blutwertkontrollen auch bei einer gut eingestellten medikamentösen Behandlung nötig.
HU – Hydroxyurea (= Hydroxycarbamid)
Hydroxyurea (Handelsnamen: Litalir, Syrea, Hydrea) gilt seit Jahrzehnten als Standardtherapie in der Behandlung von MPN-Erkrankungen. Bei HU handelt es sich um ein sogenanntes Zellteilungsgift (Zytostatikum), das die Funktion des Knochenmarks einschränkt und so die Zahl der Blutzellen reduziert. HU wirkt aber nicht nur auf die Thrombozyten, sondern hemmt ebenso die Produktion der weißen Blutkörperchen (Leukozyten) und der roten Blutkörperchen (Erythrozyten). Sind die Erythrozyten allerdings schon rückläufig und zeigt sich eine Anämie, kann diese durch HU noch verstärkt werden. Hier ist der Nutzen von HU abzuwägen und die Dosierung gegebenenfalls anzupassen. Nach dem Absetzen der Therapie steigen die Blutwerte in der Regel sehr schnell wieder an.
Gegenanzeigen
HU sollte bei jüngeren Betroffenen wegen des Einflusses von Zytostatika auf die Keimbahn (Eizellen und Spermien) und der Nebenwirkungen auf Haut und Schleimhäute zurückhaltend eingesetzt werden. Unter HU dürfen Kinder weder gezeugt noch empfangen werden, HU ist in der Schwangerschaft und Stillzeit kontraindiziert. Bei etwa fünf bis zehn Prozent der mit HU behandelten Personen bleibt die gewünschte zellreduzierende Wirkung aus (Resistenz).
„Der Übergang einer ET in ein MDS/akute Leukämie ist selten. Da medikamentöse Therapien wie Hydroxyharnstoff oder Busulfan das Risiko eines Überganges möglicherweise erhöhen, sollten sie mit Vorsicht angewendet werden.“ (aus den ET-Leitlinien der DGHO)
Einnahmeform
Tabletten beziehungsweise Kapseln (à 500 mg), in der Regel täglich
Mögliche Nebenwirkungen
Es können leichter Haarausfall und Hautveränderungen auftreten. Betroffene sollten intensive Sonneneinstrahlung meiden beziehungsweise auf einen angemessenen Sonnenschutz achten.
In einigen Fällen treten als Folge der HU-Einnahme Unterschenkelgeschwüre auf, das Medikament sollte dann sofort abgesetzt werden. Die Geschwüre bilden sich danach in der Regel wieder zurück.
Die längerfristige Behandlung mit HU geht mit einem erhöhten Risiko für Tumoren der Haut einher, Insbesondere ET-Erkrankte mit Hautkrebsvorstufen (z. B. Aktinische Keratosen) oder bösartigen Hauttumoren (z. B. Basaliome, Plattenepithelkarzinome) sollten sich regelmäßig in einer dermatologischen Praxis vorstellen, ein jährliches Hautscreening wird allen, die HU nehmen, empfohlen.
Anagrelid (Handelsnamen: Xagrid, Thromboreductin, verschiedene Generika) senkt selektiv die erhöhte Thrombozytenzahl. Die Substanz hemmt die Reifung der Megakaryozyten und schränkt damit die Neubildung von Thrombozyten ein, sodass ihre Anzahl im Blut sinkt. Es wirkt weitgehend plättchenspezifisch und beeinflusst nicht die Bildung anderer Blutzellen. Anagrelid ist als Einzeltherapie für die ET geeignet und zugelassen.
Die Effektivität, mit der Anagrelid die Thrombozytenzahl senkt, ist mit der von HU vergleichbar. Auch bei dieser Behandlung steigen die Thrombozyten nach Absetzen der Therapie im Allgemeinen schnell wieder an.
Die Ansprechrate bei Erkrankten mit MPN in der Literatur lag durchschnittlich bei etwas über 80%, erreicht aber in manchen Studien deutlich mehr als 90%.
Ein abruptes Absetzen von Anagrelid kann in Einzelfällen zu thrombotischen Komplikationen führen, weshalb es bei einem Therapiewechsel ausgeschlichen bzw. überlappend mit einer neuen Therapie eingesetzt werden sollte.
Wichtig zu wissen:
Studien haben gezeigt, dass unter der Kombination von Anagrelid und ASS die Blutungsneigung zunimmt. Daher sollten Betroffene – von begründeten Ausnahmefällen abgesehen – die beiden Medikamente insbesondere zu Beginn der Therapie bei hohen Thrombozytenwerten nicht parallel einnehmen. Dieses gilt es im Verlauf der Therapie immer wieder abzuwägen.
Einnahmeform
Tabletten beziehungsweise Kapseln (à 0,5 mg), auf 2 Dosen verteilt, täglich
Mögliche Nebenwirkungen
sind Kopfschmerzen, Durchfälle, Ödem-Bildung, Schwindel und Herzrasen. Das Nebenwirkungsrisiko sinkt deutlich, wenn der Einstieg in die Therapie mit einer geringen Dosierung erfolgt, die nach und nach gesteigert wird (Einschleichen).
Wichtig zu wissen:
Betroffene, die unter Herzrhythmusstörungen und/oder Herzinsuffizienz leiden, sollten Anagrelid nur nach sorgfältiger kardiologischer Untersuchung und einer strengen Risiko-Nutzen-Abwägung einnehmen. Der Wirkstoff führte in der Vergangenheit in Einzelfällen zu schwerwiegenden Herzproblemen, wovon in seltenen Fällen auch junge Patienten ohne Vorerkrankungen am Herzen betroffen waren.
Symptomorientierte Therapien
Die Entdeckung der JAK2-Mutation hat nicht nur das Verständnis für die MPN verbessert, sondern stellt die Basis für neue Therapieoptionen, die sogenannten JAK-Inhibitoren dar. Diese können die Aktivierung von Signalwegen durch alle relevanten Treibermutationen (JAK2, MPL, CALR) hemmen und somit zielgerichtet wirken.
Ruxolitinib (Handelsname Jakavi), ein für die PV und PMF zugelassener, sogenannter JAK-Inhibitor, ist auch bei ET wirksam, aber nicht für diese MPN-Form zugelassen. Mit Fedratinib (Handelsname Inrebic) sowie Momelotinib (Handelsname Omjjara) stehen inzwischen weitere JAK-Inhibitoren für die Behandlung der Myelofibrose zur Verfügung, die das Behandlungsspektrum prinzipiell erweitern, aber ebenfalls für die ET nicht zugelassen sind.
Erwähnenswert sind zudem neuere Entwicklungen, die für Träger der Calretikulin-Mutation langfristig interessant sein können. Derzeit laufen zwei Studien mit Antikörpertherapien bei Calretikulin mutierter MPN in Phase I/II Studien mit bislang erfreulichen Ergebnissen (Stand 2026).
Wichtig zu wissen:
Es besteht ein Unterschied zwischen Indikation und Zulassung. Während die Indikation für die Therapie-Entscheidung eine wichtige Rolle spielt, ist die Zulassung relevant für die Kostenerstattung der Therapie durch die Krankenkassen.
Bei klarer Indikation für eine zulassungsüberschreitende Therapie kann eine Kostenübernahme bei der Kasse beantragt werden. Von ärztlicher Seite wird eine Verordnung häufig aus guten Gründen gescheut, um mögliche Regress-Forderungen zu vermeiden. Ein Kostenübernahmeantrag kann dann über ein Referenzzentrum z.B. im Rahmen einer Zweitmeinung erfolgen.
Für seltene Erkrankungen oder bei einer geringen Zahl von Betroffenen werden oft keine Zulassungsstudien erfolgen, so dass die Leitlinien auch immer zulassungsüberschreitende Medikamente (Off-Label-Use) benennen, welche indiziert und wirksam sind.
Informationen zum Einsatz nicht zugelassener Arzneimittel finden sich auf der Webseite der Deutschen Leukämie- und Lymphomhilfe, Info-Blatt: Therapie Off-Label-Use, No-Label-Use, Compassionate-Use – was bedeutet das für den Patienten?
Informationen zu den Wirkstoffen Ruxolitinib, Fedratinib und Momelotinib finden Sie unter PMF/Therapieoptionen.
Interferon-alpha (INF)
Pegyliertes Interferon-alpha (Handelsnamen: Pegasys, Besremi) ist ein hormonähnlicher Botenstoff (Zytokin). Er bindet an Zellrezeptoren und kann die Produktion von Blutzellen im Knochenmark bei ET und anderen MPN verlangsamen und damit die gesteigerten Zellzahlen im peripheren Blut absenken. Interferon-alpha scheint nicht nur auf Wachstumsfaktoren und andere Botenstoffe, die Zytokine, zu wirken, sondern auch faserbildende Zellen zu hemmen, wodurch die Wahrscheinlichkeit des Übergangs einer ET in eine Myelofibrose reduziert werden kann.
Interferon-alpha Pegasys ist in Deutschland seit 2024 für die Behandlung der ET zugelassen. Für das für die PV zugelassene Interferon-alpha Besremi wurde eine Zulassung in den USA für die Behandlung der ET beantragt. Sie wird gegen Ende 2026 erwartet. Sollte diese erfolgen, ist in der Regel mit einigen Monaten Verzögerung auch mit einer Zulassung in Europa zu rechnen.
Wichtig zu wissen:
Für ET-Patientinnen, die schwanger sind oder einen Kinderwunsch haben und eine zellreduzierende Behandlung benötigen, ist Interferon-alpha aktuell das Mittel der Wahl und wird in den Leitlinien der DGHO empfohlen.
Gegenanzeigen
Interferon sollte bei Betroffenen mit bestimmten Schilddrüsenerkrankungen (z. B. Hashimoto-Thyreoiditis) nur dann verordnet werden, wenn diese mit entsprechenden Medikamenten kontrolliert werden können.
Nicht selten treten neurologische und psychische Probleme auf, bei Erkrankten mit psychischen Vorerkrankungen wie Depressionen können sich diese deutlich verstärken. Diese Problematik muss unbedingt beachtet werden. Eventuell kann es sinnvoll sein, vor und unter Therapie regelmäßig eine psychiatrische oder psychologische Beratung in Anspruch zu nehmen. Nach Absetzen bzw. Dosisreduktion von Interferon gehen diese Nebenwirkungen häufig schnell wieder zurück.
Patienten, die vor Kurzem einen Herzinfarkt oder Schlaganfall hatten, sollten auf die Anwendung von Interferon zumindest vorerst verzichten. Vorsichtiger Einsatz ist ebenfalls bei schwerer Leber- oder Nierenerkrankung und bei Autoimmunerkrankungen angeraten.
Einnahmeform
Ähnlich wie Insulin bei Diabetikern, muss Interferon mit Fertigspritzen selbst unter die Haut (subkutan) gespritzt werden, bei der pegylierten Form einmal wöchentlich oder alle 14 Tage, im Behandlungsverlauf manchmal auch in noch größeren Abständen.
Durch die Pegylierung des Wirkstoffs bei den heute üblichen Präparaten wie Pegasys oder Besremi wird der Abbau des Medikaments im Körper verlangsamt, sodass in größeren Abständen gespritzt werden kann (z. B. alle 7-14 Tage statt alle drei Tage). Zusätzlich kommt es zu einer relativ konstanten Wirkstoffkonzentration im Körper, Nebenwirkungen können so reduziert werden.
Mögliche Nebenwirkungen
Sehr häufige Nebenwirkungen von (pegylierten) Interferonen, die mehr als eine von zehn Personen betreffen können, sind niedrige Spiegel der weißen Blutkörperchen (Leukozyten) und der Blutplättchen (Thrombozyten).
Mögliche weitere Nebenwirkungen sind Muskel- und Gelenkschmerzen, Müdigkeit, grippeähnliche Symptome nach der Injektion (Fieber, Muskel- und Knochenschmerzen, Schüttelfrost, Übelkeit, Durchfall), Rötungen oder Reizungen an der Injektionsstelle, Haarausfall, Hautreaktionen, Depressionen, Gewichtsverlust, Konzentrationsstörungen, erhöhte Leberwerte im Blut und Störungen der Schilddrüsenfunktion. Da Interferon das Immunsystem stimuliert, können in einigen Fällen Autoimmunerkrankungen ausgelöst oder bereits bestehende verstärkt werden.
Ein Einschleichen der Therapie mit niedrigen, langsam ansteigenden Dosierungen kann die Nebenwirkungen mildern. Nach einer Gewöhnungsphase gehen diese in der Regel deutlich zurück.
Gibt es noch andere Behandlungsmethoden?
Komplementärmedizinische Verfahren sind nur in Ergänzung zur schulmedizinischen Behandlung zu sehen. Sie können die Begleitsymptomatik verbessern und damit die Lebensqualität erhöhen.
Einen verantwortungsvollen Therapeuten erkennt man unter anderem daran, dass er nicht vorgibt, die ET mit alternativen Methoden heilen zu können.
Mehr erfahren Sie unter Lebensqualität trotz MPN.
Es finden immer wieder weltweit klinische Studien zur Erforschung neuer Therapieansätze für die ET statt. Wenn sich eine neue Behandlungsmethode als wirksam und verträglich herausstellt, kann diese Therapie Eingang in die Therapieleitlinien finden.
Regelmäßige Kontrollen sind wichtig
Die Behandlungsleitlinien der Deutschen Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO) empfehlen folgende Untersuchungen, um den Verlauf der ET dauerhaft zu kontrollieren:
- Blutuntersuchung: Die Abstände können je nach Schwere der Erkrankung und Therapie zwischen wenigen Tagen (z.B. bei Therapieeinstieg) und mehreren Monaten (bei stabilem Verlauf) variieren
- eine halbjährliche umfassende Untersuchung mit einem ausführlichen ärztlichen Gespräch, um den Krankheitsverlauf, mögliche Komplikationen und Therapienebenwirkungen zu überwachen
- etwa jährlicher Ultraschall des Oberbauchs, vor allem wegen möglicher Milzvergrößerung
Eine erneute Untersuchung des Knochenmarks ist nur nötig, wenn sich das Befinden oder die Blutwerte gravierend verändern. Dies kann ein Hinweis darauf sein, dass die Krankheit voranschreitet. Vor einer geplanten Therapieumstellung kann eine Knochenmarkbiopsie ebenfalls sinnvoll sein.
Sämtliche Empfehlungen der DGHO dienen lediglich als Orientierung. Ausschlaggebend für die Häufigkeit von bestimmten Untersuchungen sind der individuelle Krankheitsverlauf und das Befinden. Im Idealfall arbeiten Ärztin und Patient in diesen Fragen zusammen.
Fazit
Keine kurative Behandlung
Trotz neuer, hoffnungsvoller Ansätze in der Medikamentenforschung gehört die ET zu den Erkrankungen, für die es derzeit keine kurative (heilende) Behandlung gibt.
Vielmehr muss man von einer medizinischen Begleitung der Krankheit sprechen, bei der die ärztliche Aufgabe primär darin besteht, mögliche Komplikationen der Blutbildungsstörung zu verhindern beziehungsweise die Symptome des Erkrankten zu behandeln.
Einzelne Therapieansätze werden aber aktuell darauf untersucht, ob sie ggf. den Verlauf der Erkrankung beeinflussen können.
Der Erhalt beziehungsweise die Wiederherstellung der Lebensqualität sollte in der Zusammenarbeit von Arzt und Patientin im Vordergrund stehen.
»Primum non nocere« – »zuallererst: Füge keinen Schaden zu.«
Einer der weltweit anerkannten Hämatologen, Dr. Jerry Le Pow Spivak aus den USA, erinnert für die Behandlung der PV an einen sehr alten ärztlichen Grundsatz (um 50 n. Chr.): »Primum non nocere« – »zuallererst: Füge keinen Schaden zu.«
Dr. Spivak legt diesen Grundsatz folgendermaßen aus: »Die Diagnose muss korrekt sein, die Therapie sollte ebenso sicher wie effektiv und die Behandlung nicht schlimmer als die Erkrankung selbst sein.« (Übersetzung Dieter Wenzel) Diese Forderung macht deutlich, dass es in der Begleitung der PV für die Ärztin keine Strategie gibt, die sich auf jeden Patienten zu jeder Zeit pauschal anwenden lässt. Ziel muss vielmehr sein, die individuell richtige Behandlung zu finden, die dem einzelnen Betroffenen am meisten nutzt und seine Lebensqualität so lange wie möglich erhält.
MPN-Register
Zusätzlich zur Erforschung neuer Medikamente ist es für eine effektive Therapie von ET-Betroffenen unverzichtbar, die Erfahrungen mit bisherigen Behandlungsstrategien systematisch zu erfassen und auszuwerten. Vor diesem Hintergrund begrüßt das mpn-netzwerk e. V. ausdrücklich die Einführung des Registers der German Study Group for MPN (GSG-MPN Register) und empfiehlt allen Betroffenen, durch ihre Teilnahme aktiv zur Vermehrung des Wissens über MPN-Erkrankungen und deren Behandlung beizutragen.
Austausch mit anderen Betroffenen
Der Austausch mit anderen Betroffenen in Selbsthilfegruppen oder -organisationen kann helfen, das Hier und Jetzt nicht zu vergessen. Das mpn-netzwerk e. V. lädt Sie daher abschließend herzlich ein, sich am Austausch im Forum sowie auf den Regional- und Jahrestreffen des Vereins zu beteiligen.
Wir danken Prof. Steffen Koschmieder, Aachen, für die fachliche Beratung und das Gegenlesen der Ursprungsfassung dieses Textes.
Quellen und Links:
- Broschüre ET, 5. Auflage, mpn-netzwerk e.V.
- Webseite DLH der Deutschen Leukämie- und Lymphomhilfe/Informationsblätter
https://www.leukaemie-hilfe.de/infothek/eigene-publikationen/infoblaetter/ - DGHO – Deutsche Gesellschaft für Hämatologie und Onkologie: Aktuelle Leitlinie für Ärzte zu Diagnose und Therapie der ET
https://www.onkopedia.com/de/onkopedia/guidelines/essentielle-oder-primaere-thrombozythaemie-et/@@guideline/html/index.html - Webseite GSG-MPN
https://www.gsg-mpn.de/gsgmpn - Mayo-Clinic
https://onlinelibrary.wiley.com/doi/10.1002/ajh.25270 - Mikroskopie-Abbildung – mit freundlicher Genehmigung aus: Torsten Haferlach, Hämatologische Erkrankungen, Atlas und diagnostisches Handbuch, Springer-Verlag Berlin Heidelberg 2020, ISBN 978-3-662-59546-6
 .
.
